publications
publications by categories in reversed chronological order.
* denotes co-first authors
2025
-
 Accurate predictions of conformational ensembles of disordered proteins with STARLINGBorna Novak*, Jeffrey M Lotthammer*, Ryan J Emenecker, and 1 more authorbioRxiv, Feb 2025
Accurate predictions of conformational ensembles of disordered proteins with STARLINGBorna Novak*, Jeffrey M Lotthammer*, Ryan J Emenecker, and 1 more authorbioRxiv, Feb 2025Intrinsically disordered proteins and regions (collectively IDRs) are found across all kingdoms of life and play critical roles in virtually every eukaryotic cellular process. In contrast to folded proteins, IDRs lack a stable 3D structure and are instead described in terms of a conformational ensemble, a collection of energetically accessible interconverting structures. This unique structural plasticity facilitates diverse molecular recognition and function; thus, a convenient way to view IDRs is through their ensembles. Here, we combine advances in physics-based force fields for IDPs with the power of modern multi-scale generative modeling to develop STARLING, an approach for the rapid and accurate prediction of IDR ensembles directly from sequence. STARLING enables ensembles of hundreds of conformers to be generated in seconds and works on GPUs and CPUs. This, in turn, dramatically lowers the barrier to the computational interrogation of IDR function through the lens of emergent biophysical properties in addition to traditional bioinformatic approaches. We evaluate STARLING’s accuracy against experimental data and offer a series of vignettes illustrating how STARLING can enable rapid hypothesis generation for IDR function or the interpretation of experimental data.
-
 Disentangling Folding from Energetic Traps in Simulations of Disordered ProteinsJeffrey M Lotthammer, and Alex S HolehouseJournal of Chemical Information and Modeling, Mar 2025
Disentangling Folding from Energetic Traps in Simulations of Disordered ProteinsJeffrey M Lotthammer, and Alex S HolehouseJournal of Chemical Information and Modeling, Mar 2025Protein conformational heterogeneity plays an essential role in a myriad of different biological processes. Extensive conformational heterogeneity is especially characteristic of intrinsically disordered proteins and protein regions (collectively IDRs), which lack a well-defined three-dimensional structure and instead rapidly exchange between a diverse ensemble of configurations. An emerging paradigm recognizes that the conformational biases encoded in IDR ensembles can play a central role in their biological function, necessitating understanding these sequence–ensemble relations. All-atom simulations have provided critical insight into our modern understanding of the solution behavior of IDRs. However, effectively exploring the accessible conformational space associated with large, heterogeneous ensembles is challenging. In particular, identifying poorly sampled or energetically trapped regions of disordered proteins in simulations often relies on qualitative assessment based on visual inspection of simulations and/or analysis data. These approaches, while convenient, run the risk of masking poorly sampled simulations. In this work, we present an algorithm for quantifying per-residue local conformational heterogeneity in protein simulations. Our work builds on prior work and compares the similarity between backbone dihedral angle distributions generated from molecular simulations in a limiting polymer model and across independent all-atom simulations. In this regime, the polymer model serves as a statistical reference model for extensive conformational heterogeneity in a real chain. Quantitative comparisons of probability vectors generated from these simulations reveal the extent of conformational sampling in a simulation, enabling us to distinguish between situations in which protein regions are well-sampled, poorly sampled, or folded. To demonstrate the effectiveness of this approach, we apply our algorithm to several toy, synthetic, and biological systems. Accurately assessing local conformational sampling in simulations of IDRs will help better quantify new enhanced sampling methods, ensure force field comparisons are equivalent, and provide confidence that conclusions drawn from simulations are robust.
- Complexes of vertebrate TMC1/2 and CIB2/3 proteins form hair-cell mechanotransduction cation channelsArnaud P J Giese, Wei-Hsiang Weng, Katie S Kindt, and 14 more authorsElife, Jan 2025
Calcium and integrin-binding protein 2 (CIB2) and CIB3 bind to transmembrane channel-like 1 (TMC1) and TMC2, the pore-forming subunits of the inner-ear mechano-electrical transduction (MET) apparatus. These interactions have been proposed to be functionally relevant across mechanosensory organs and vertebrate species. Here, we show that both CIB2 and CIB3 can form heteromeric complexes with TMC1 and TMC2 and are integral for MET function in mouse cochlea and vestibular end organs as well as in zebrafish inner ear and lateral line. Our AlphaFold 2 models suggest that vertebrate CIB proteins can simultaneously interact with at least two cytoplasmic domains of TMC1 and TMC2 as validated using nuclear magnetic resonance spectroscopy of TMC1 fragments interacting with CIB2 and CIB3. Molecular dynamics simulations of TMC1/2 complexes with CIB2/3 predict that TMCs are structurally stabilized by CIB proteins to form cation channels. Overall, our work demonstrates that intact CIB2/3 and TMC1/2 complexes are integral to hair-cell MET function in vertebrate mechanosensory epithelia.

2024
-
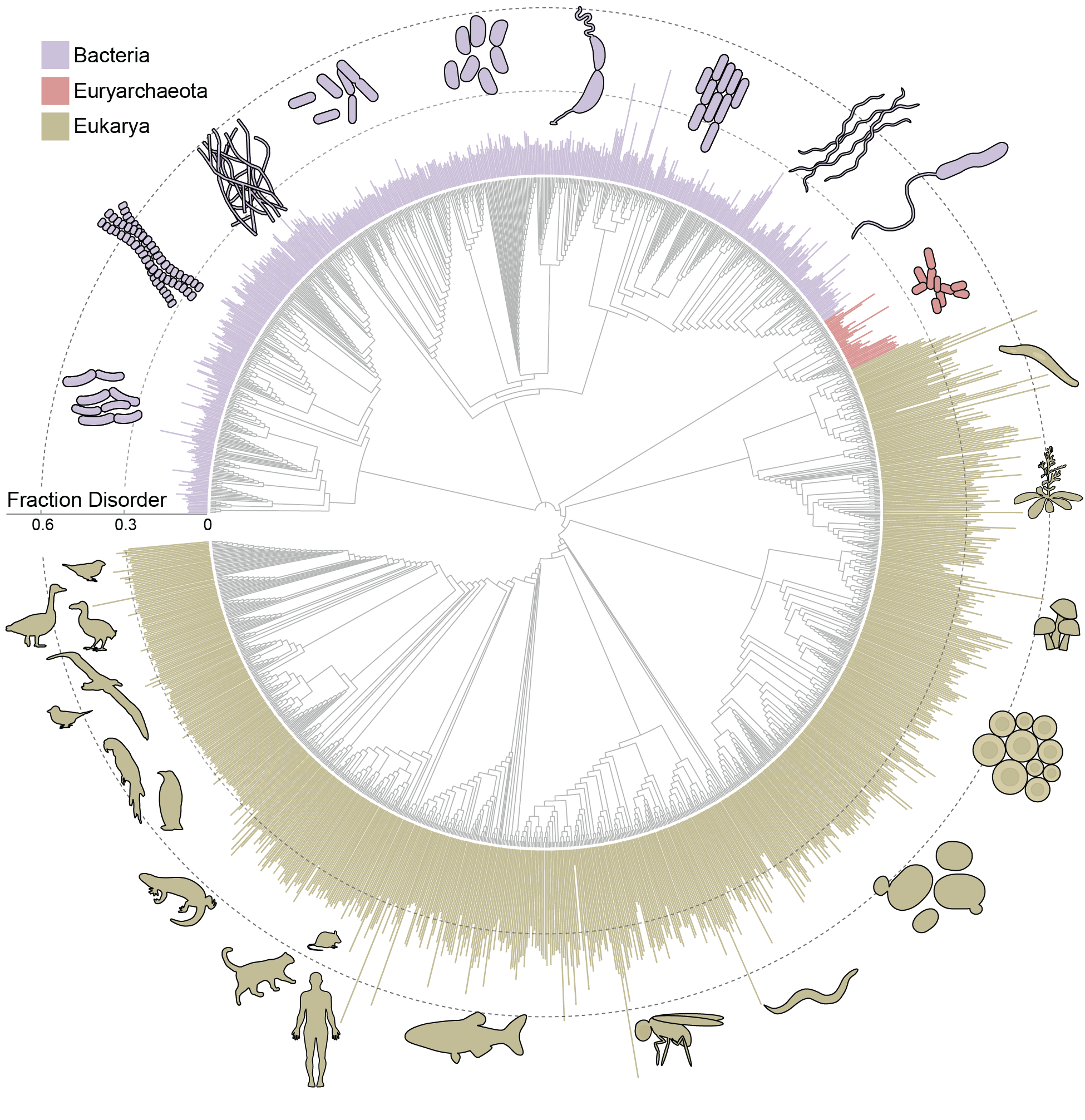 Metapredict enables accurate disorder prediction across the Tree of LifeJeffrey M Lotthammer, Jorge Hernández-García, Daniel Griffith, and 3 more authorsbioRxiv, Nov 2024
Metapredict enables accurate disorder prediction across the Tree of LifeJeffrey M Lotthammer, Jorge Hernández-García, Daniel Griffith, and 3 more authorsbioRxiv, Nov 2024Intrinsically disordered proteins and protein regions (collectively IDRs) are critical in numerous cellular processes. To understand how IDRs facilitate function, we need tools to accurately and rapidly identify them from sequence. While many methods for disorder prediction exist, we are currently limited by throughput and accuracy for evolutionary scale analyses. To bridge this gap, we developed metapredict V3, an updated version of our disorder predictor that enables evolutionary-scale disorder prediction. Metapredict V3 enables proteome-scale prediction with state-of-the-art accuracy in seconds and was developed with a focus on usability. It is distributed as a web server, Python software package, command-line interface, and Google Colab notebook. Here, we leverage the accuracy and throughput of metapredict V3 to predict disorder for over 20,000 proteomes to evaluate the prevalence of disorder across the kingdoms of life.
-
 Rhythmidia: A modern tool for circadian period analysis of filamentous fungiAlex T Keeley, Jeffrey M Lotthammer, and Jacqueline F PelhamPLoS Comput. Biol., Aug 2024
Rhythmidia: A modern tool for circadian period analysis of filamentous fungiAlex T Keeley, Jeffrey M Lotthammer, and Jacqueline F PelhamPLoS Comput. Biol., Aug 2024Circadian rhythms are ubiquitous across the kingdoms of life and serve important roles in regulating physiology and behavior at many levels. These rhythms occur in 24-hour cycles and are driven by a core molecular oscillator. Circadian timekeeping enables organisms to anticipate daily changes by timing their growth and internal processes. Neurospora crassa is a model organism with a long history in circadian biology, having conserved eukaryotic clock properties and observable circadian phenotypes. A core approach for measuring circadian function in Neurospora is to follow daily oscillations in the direction of growth and spore formation along a thin glass tube (race tube). While leveraging robust phenotypic readouts is useful, interpreting the outputs of large-scale race tube experiments by hand can be time-consuming and prone to human error. To provide the field with an efficient tool for analyzing race tubes, we present Rhythmidia, a graphical user interface (GUI) tool written in Python for calculating circadian periods and growth rates of Neurospora. Rhythmidia is open source, has been benchmarked against the current state-of-the-art, and is easily accessible on GitHub.
- Protein surface chemistry encodes an adaptive tolerance to desiccationPaulette Sofía Romero-Pérez, Haley M Moran, Azeem Horani, and 14 more authorsbioRxiv, Oct 2024
Cellular desiccation - the loss of nearly all water from the cell - is a recurring stress in an increasing number of ecosystems that can drive protein unfolding and aggregation. For cells to survive, at least some of the proteome must resume function upon rehydration. Which proteins tolerate desiccation, and the molecular determinants that underlie this tolerance, are largely unknown. Here, we apply quantitative and structural proteomic mass spectrometry to show that certain proteins possess an innate capacity to tolerate rehydration following extreme water loss. Structural analysis points to protein surface chemistry as a key determinant for desiccation tolerance, which we test by showing that rational surface mutants can convert a desiccation sensitive protein into a tolerant one. Desiccation tolerance also has strong overlap with cellular function, with highly tolerant proteins responsible for production of small molecule building blocks, and intolerant proteins involved in energy-consuming processes such as ribosome biogenesis. As a result, the rehydrated proteome is preferentially enriched with metabolite and small molecule producers and depleted of some of the cell’s heaviest consumers. We propose this functional bias enables cells to kickstart their metabolism and promote cell survival following desiccation and rehydration.
-
 Direct prediction of intermolecular interactions driven by disordered regionsGarrett M Ginell, Ryan J Emenecker, Jeffrey M Lotthammer, and 2 more authorsbioRxiv, Jun 2024
Direct prediction of intermolecular interactions driven by disordered regionsGarrett M Ginell, Ryan J Emenecker, Jeffrey M Lotthammer, and 2 more authorsbioRxiv, Jun 2024Intrinsically disordered regions (IDRs) are critical for a wide variety of cellular functions, many of which involve interactions with partner proteins. Molecular recognition is typically considered through the lens of sequence-specific binding events. However, a growing body of work has shown that IDRs often interact with partners in a manner that does not depend on the precise order of the amino acid order, instead driven by complementary chemical interactions leading to disordered bound-state complexes. Despite this emerging paradigm, we lack tools to describe, quantify, predict, and interpret these types of structurally heterogeneous interactions from the underlying amino acid sequences. Here, we repurpose the chemical physics developed originally for molecular simulations to develop an approach for predicting intermolecular interactions between IDRs and partner proteins. Our approach enables the direct prediction of phase diagrams, the identification of chemically-specific interaction hotspots on IDRs, and a route to develop and test mechanistic hypotheses regarding IDR function in the context of molecular recognition. We use our approach to examine a range of systems and questions to highlight its versatility and applicability.
-
 Direct prediction of intrinsically disordered protein conformational properties from sequenceJeffrey M Lotthammer*, Garrett M Ginell*, Daniel Griffith*, and 2 more authorsNature Methods, Mar 2024
Direct prediction of intrinsically disordered protein conformational properties from sequenceJeffrey M Lotthammer*, Garrett M Ginell*, Daniel Griffith*, and 2 more authorsNature Methods, Mar 2024Intrinsically disordered regions (IDRs) are ubiquitous across all domains of life and play a range of functional roles. While folded domains are generally well described by a stable three-dimensional structure, IDRs exist in a collection of interconverting states known as an ensemble. This structural heterogeneity means that IDRs are largely absent from the Protein Data Bank, contributing to a lack of computational approaches to predict ensemble conformational properties from sequence. Here we combine rational sequence design, large-scale molecular simulations and deep learning to develop ALBATROSS, a deep-learning model for predicting ensemble dimensions of IDRs, including the radius of gyration, end-to-end distance, polymer-scaling exponent and ensemble asphericity, directly from sequences at a proteome-wide scale. ALBATROSS is lightweight, easy to use and accessible as both a locally installable software package and a point-and-click-style interface via Google Colab notebooks. We first demonstrate the applicability of our predictors by examining the generalizability of sequence-ensemble relationships in IDRs. Then, we leverage the high-throughput nature of ALBATROSS to characterize the sequence-specific biophysical behavior of IDRs within and between proteomes.
-
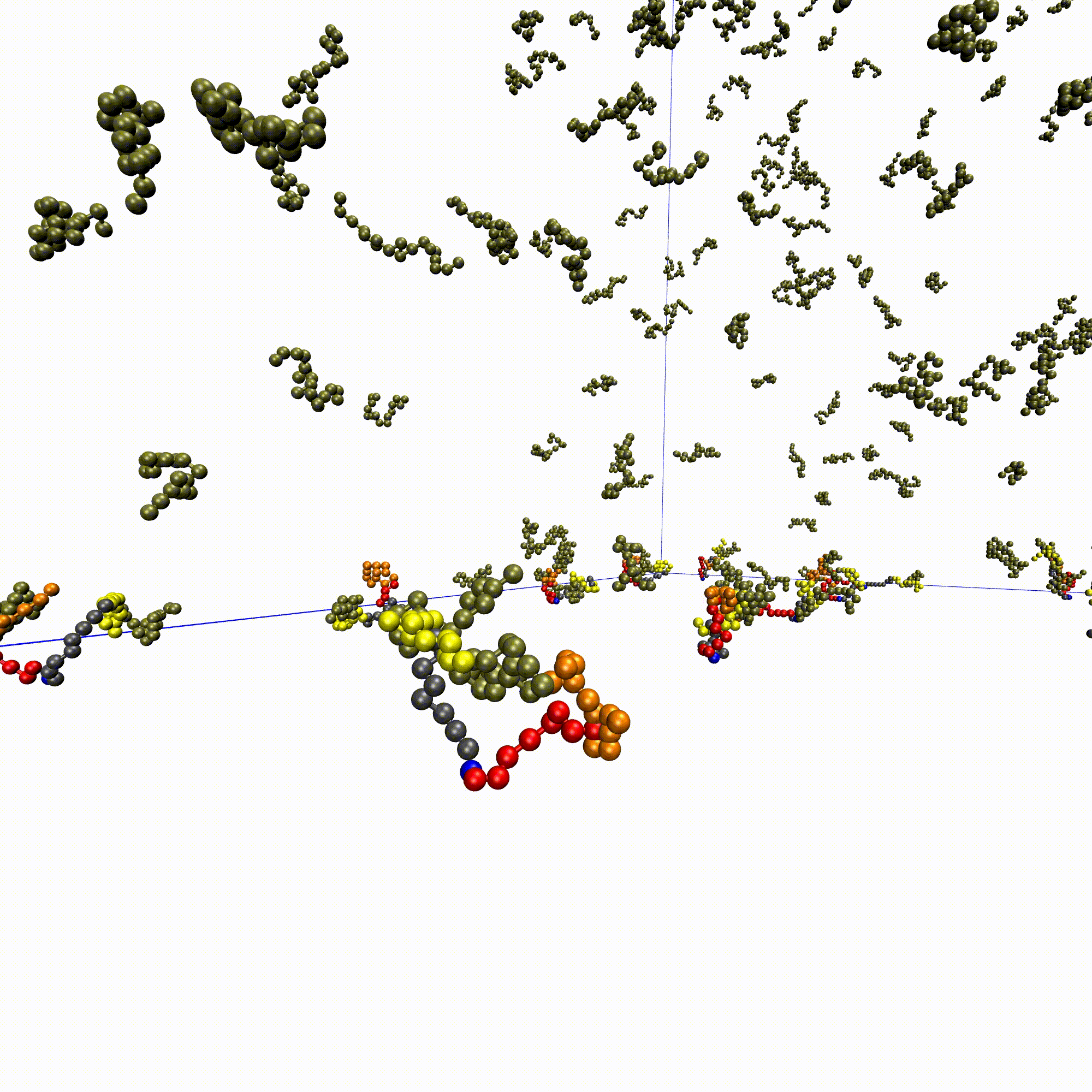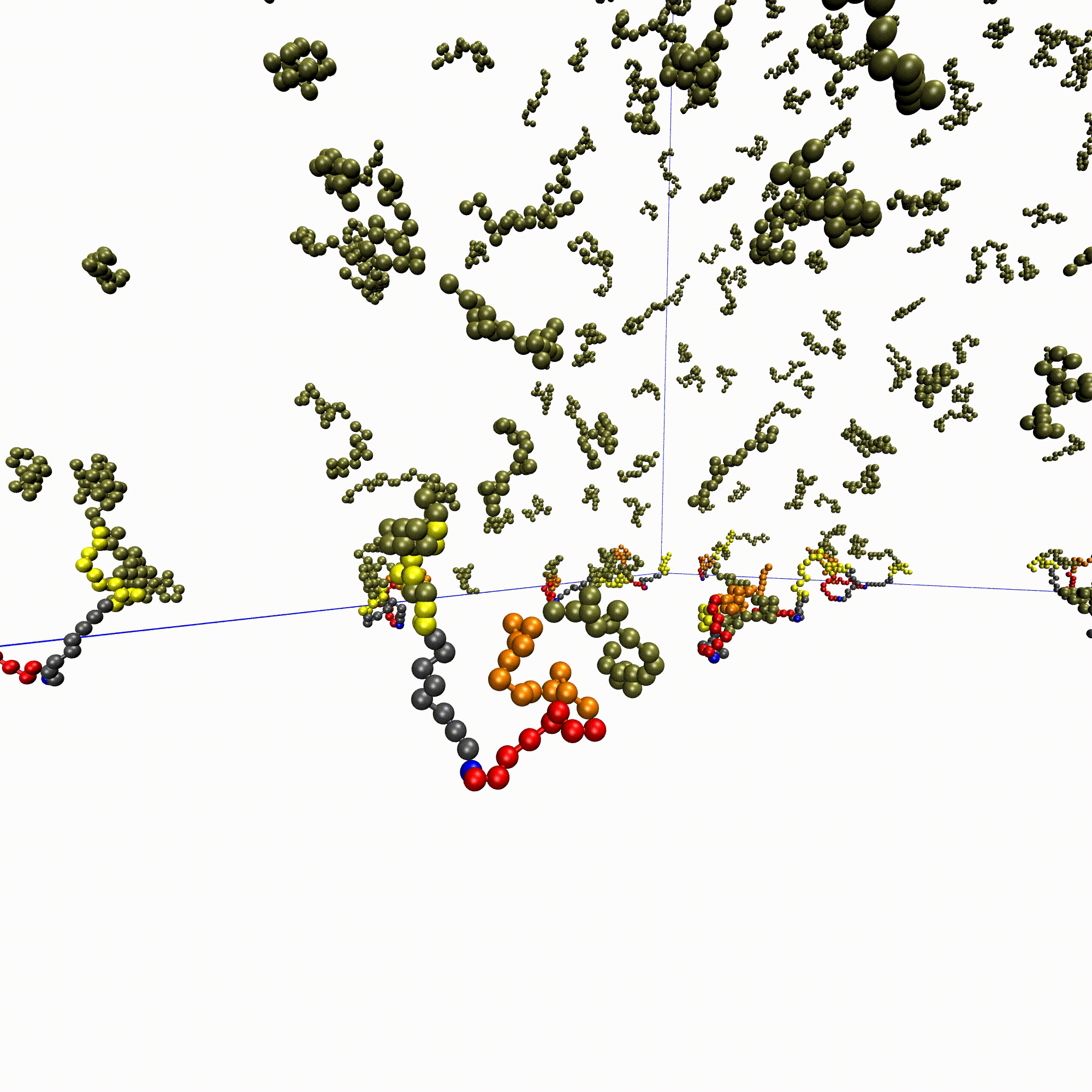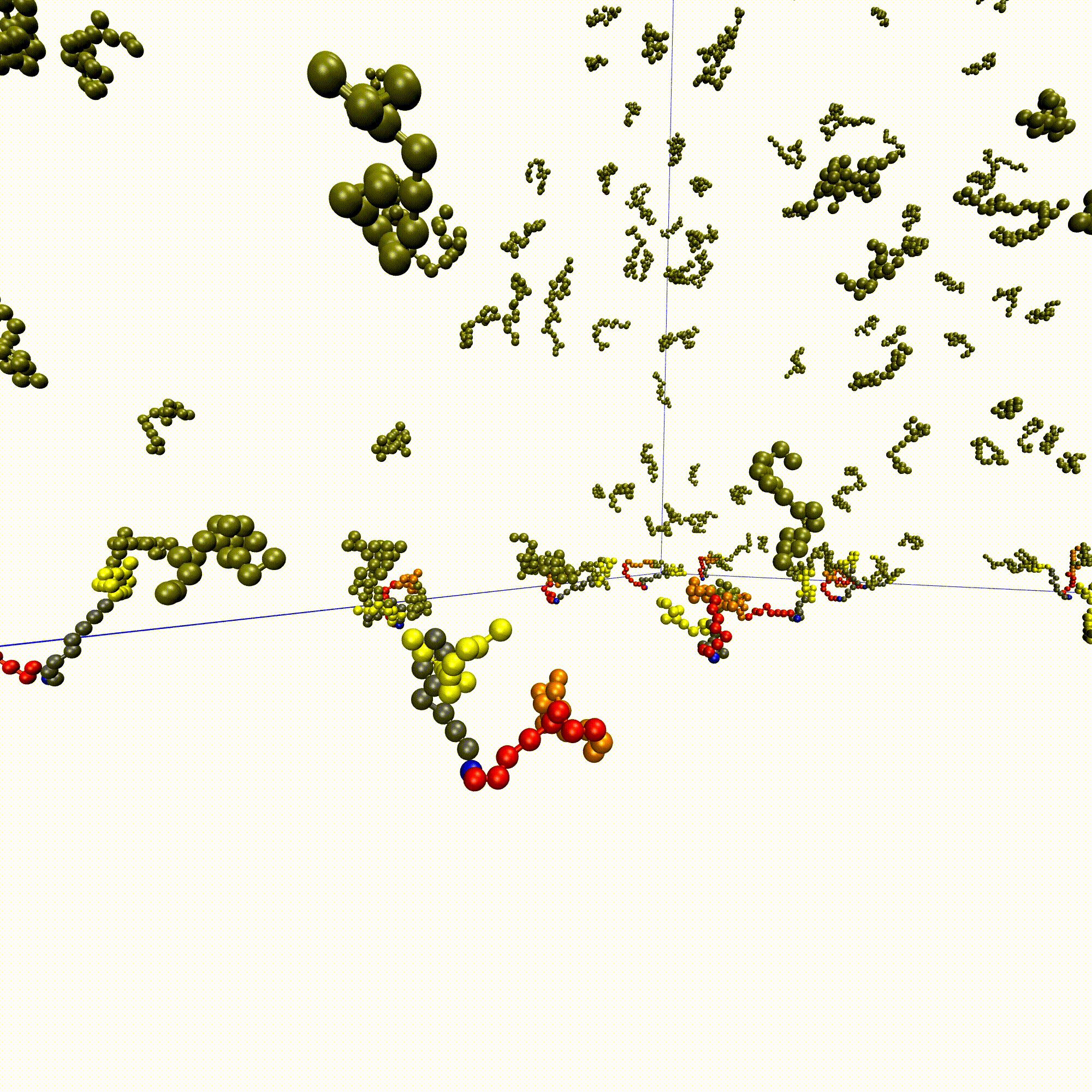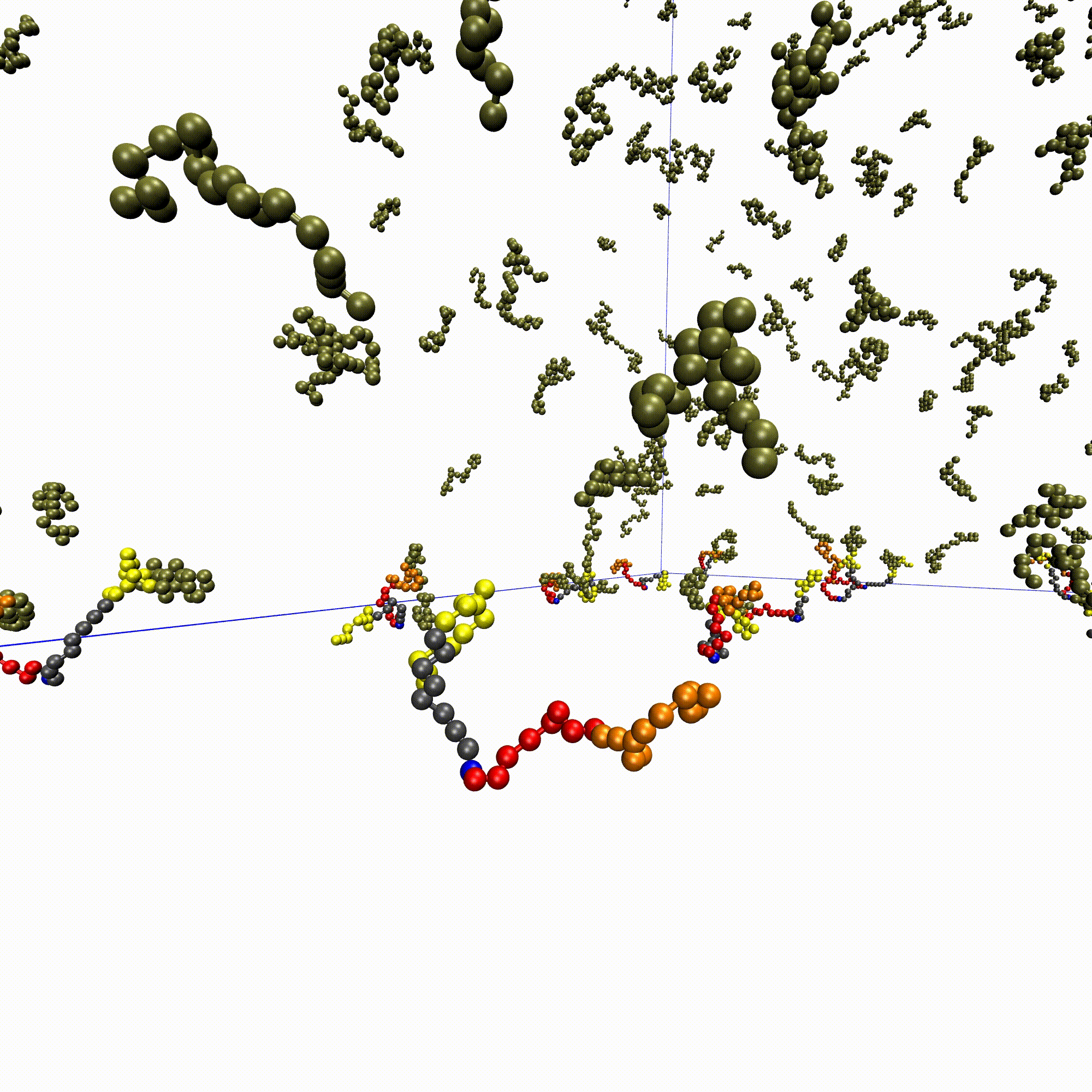 High-throughput affinity measurements of direct interactions between activation domains and co-activatorsNicole DelRosso, Peter H Suzuki, Daniel Griffith, and 7 more authorsbioRxiv, Aug 2024
High-throughput affinity measurements of direct interactions between activation domains and co-activatorsNicole DelRosso, Peter H Suzuki, Daniel Griffith, and 7 more authorsbioRxiv, Aug 2024Sequence-specific activation by transcription factors is essential for gene regulation1,2. Key to this are activation domains, which often fall within disordered regions of transcription factors3,4 and recruit co-activators to initiate transcription5. These interactions are difficult to characterize via most experimental techniques because they are typically weak and transient6,7. Consequently, we know very little about whether these interactions are promiscuous or specific, the mechanisms of binding, and how these interactions tune the strength of gene activation. To address these questions, we developed a microfluidic platform for expression and purification of hundreds of activation domains in parallel followed by direct measurement of co-activator binding affinities (STAMMPPING, for Simultaneous Trapping of Affinity Measurements via a Microfluidic Protein-Protein INteraction Generator). By applying STAMMPPING to quantify direct interactions between eight co-activators and 204 human activation domains (>1,500 K ds), we provide the first quantitative map of these interactions and reveal 334 novel binding pairs. We find that the metazoan-specific co-activator P300 directly binds >100 activation domains, potentially explaining its widespread recruitment across the genome to influence transcriptional activation. Despite sharing similar molecular properties (e.g. enrichment of negative and hydrophobic residues), activation domains utilize distinct biophysical properties to recruit certain co-activator domains. Co-activator domain affinity and occupancy are well-predicted by analytical models that account for multivalency, and in vitro affinities quantitatively predict activation in cells with an ultrasensitive response. Not only do our results demonstrate the ability to measure affinities between even weak protein-protein interactions in high throughput, but they also provide a necessary resource of over 1,500 activation domain/co-activator affinities which lays the foundation for understanding the molecular basis of transcriptional activation.




2023
- Functional divergence of the sarcomeric myosin, MYH7b, supports species-specific biological rolesLindsey A Lee, Samantha K Barrick, Artur Meller, and 8 more authorsJ. Biol. Chem., Jan 2023
Myosin heavy chain 7b (MYH7b) is an evolutionarily ancient member of the sarcomeric myosin family, which typically supports striated muscle function. However, in mammals, alternative splicing prevents MYH7b protein production in cardiac and most skeletal muscles and limits expression to a subset of specialized muscles and certain nonmuscle environments. In contrast, MYH7b protein is abundant in python cardiac and skeletal muscles. Although the MYH7b expression pattern diverges in mammals versus reptiles, MYH7b shares high sequence identity across species. So, it remains unclear how mammalian MYH7b function may differ from that of other sarcomeric myosins and whether human and python MYH7b motor functions diverge as their expression patterns suggest. Thus, we generated recombinant human and python MYH7b protein and measured their motor properties to investigate any species-specific differences in activity. Our results reveal that despite having similar working strokes, the MYH7b isoforms have slower actin-activated ATPase cycles and actin sliding velocities than human cardiac β-MyHC. Furthermore, python MYH7b is tuned to have slower motor activity than human MYH7b because of slower kinetics of the chemomechanical cycle. We found that the MYH7b isoforms adopt a higher proportion of myosin heads in the ultraslow, super-relaxed state compared with human cardiac β-MyHC. These findings are supported by molecular dynamics simulations that predict MYH7b preferentially occupies myosin active site conformations similar to those observed in the structurally inactive state. Together, these results suggest that MYH7b is specialized for slow and energy-conserving motor activity and that differential tuning of MYH7b orthologs contributes to species-specific biological roles.
- Drug specificity and affinity are encoded in the probability of cryptic pocket opening in myosin motor domainsArtur Meller, Jeffrey M Lotthammer, Louis G Smith, and 7 more authorsElife, Jan 2023
The design of compounds that can discriminate between closely related target proteins remains a central challenge in drug discovery. Specific therapeutics targeting the highly conserved myosin motor family are urgently needed as mutations in at least six of its members cause numerous diseases. Allosteric modulators, like the myosin-II inhibitor blebbistatin, are a promising means to achieve specificity. However, it remains unclear why blebbistatin inhibits myosin-II motors with different potencies given that it binds at a highly conserved pocket that is always closed in blebbistatin-free experimental structures. We hypothesized that the probability of pocket opening is an important determinant of the potency of compounds like blebbistatin. To test this hypothesis, we used Markov state models (MSMs) built from over 2 ms of aggregate molecular dynamics simulations with explicit solvent. We find that blebbistatin’s binding pocket readily opens in simulations of blebbistatin-sensitive myosin isoforms. Comparing these conformational ensembles reveals that the probability of pocket opening correctly identifies which isoforms are most sensitive to blebbistatin inhibition and that docking against MSMs quantitatively predicts blebbistatin binding affinities (R2=0.82). In a blind prediction for an isoform (Myh7b) whose blebbistatin sensitivity was unknown, we find good agreement between predicted and measured IC50s (0.67 μM vs. 0.36 μM). Therefore, we expect this framework to be useful for the development of novel specific drugs across numerous protein targets.
- Divergent Molecular Phenotypes in Point Mutations at the Same Residue in Beta-Myosin Heavy Chain Lead to Distinct CardiomyopathiesSarah J Lehman, Artur Meller, Shahlo O Solieva, and 7 more authorsbioRxiv, Jul 2023
In genetic cardiomyopathies, a frequently described phenomenon is how similar mutations in one protein can lead to discrete clinical phenotypes. One example is illustrated by two mutations in beta myosin heavy chain (β-MHC) that are linked to hypertrophic cardiomyopathy (HCM) (Ile467Val, I467V) and left ventricular non-compaction (LVNC) (Ile467Thr, I467T). To investigate how these missense mutations lead to independent diseases, we studied the molecular effects of each mutation using recombinant human β-MHC Subfragment 1 (S1) in in vitro assays. Both HCM-I467V and LVNC-I467T S1 mutations exhibited similar mechanochemical function, including unchanged ATPase and enhanced actin velocity but had opposing effects on the super-relaxed (SRX) state of myosin. HCM-I467V S1 showed a small reduction in the SRX state, shifting myosin to a more actin-available state that may lead to the “gain-of-function” phenotype commonly described in HCM. In contrast, LVNC-I467T significantly increased the population of myosin in the ultra-slow SRX state. Interestingly, molecular dynamics simulations reveal that I467T allosterically disrupts interactions between ADP and the nucleotide-binding pocket, which may result in an increased ADP release rate. This predicted change in ADP release rate may define the enhanced actin velocity measured in LVNC-I467T, but also describe the uncoupled mechanochemical function for this mutation where the enhanced ADP release rate may be sufficient to offset the increased SRX population of myosin. These contrasting molecular effects may lead to contractile dysregulation that initiates LVNC-associated signaling pathways that progress the phenotype. Together, analysis of these mutations provides evidence that phenotypic complexity originates at the molecular level and is critical to understanding disease progression and developing therapies.
2021
-
 In Silico Electrophysiology of Inner-Ear Mechanotransduction Channel TMC1 ModelsSanket Walujkar, Jeffrey M Lotthammer, Collin R Nisler, and 3 more authorsbioRxiv, Sep 2021
In Silico Electrophysiology of Inner-Ear Mechanotransduction Channel TMC1 ModelsSanket Walujkar, Jeffrey M Lotthammer, Collin R Nisler, and 3 more authorsbioRxiv, Sep 2021Inner-ear sensory hair cells convert mechanical stimuli from sound and head movements into electrical signals during mechanotransduction. Identification of all molecular components of the inner-ear mechanotransduction apparatus is ongoing; however, there is strong evidence that TMC1 and TMC2 are pore-forming subunits of the complex. We present molecular dynamics simulations that probe ion conduction of TMC1 models built based on two different structures of related TMEM16 proteins. Unlike most channels, the TMC1 models do not show a central pore. Instead, simulations of these models in a membrane environment at various voltages reveal a peripheral permeation pathway that is exposed to lipids and that shows cation permeation at rates comparable to those measured in hair cells. Furthermore, our analyses suggest that TMC1 gating mechanisms involve protein conformational changes and tension-induced lipid-mediated pore widening. These results provide insights into ion conduction and activation mechanisms of hair-cell mechanotransduction channels essential for hearing and balance.
